INFLAMMATION¶
RAPID & DURABLE
DISEASE CONTROL IN AS
Not an actual AS or nr-axSpA patient
RINVOQ achieved its primary endpoint of ASAS40 at Week 14
with responses observed at Week 4, up to 2 years.1-3
INDICATION
RINVOQ is indicated for the treatment of adults with active ankylosing spondylitis (AS) who have had an inadequate response or intolerance to one or more tumor necrosis factor (TNF) blockers.
Limitations of Use: RINVOQ is not recommended for use in combination with other Janus kinase (JAK) inhibitors, biologic disease-modifying antirheumatic drugs (bDMARDs), or with potent immunosuppressants such as azathioprine and cyclosporine.
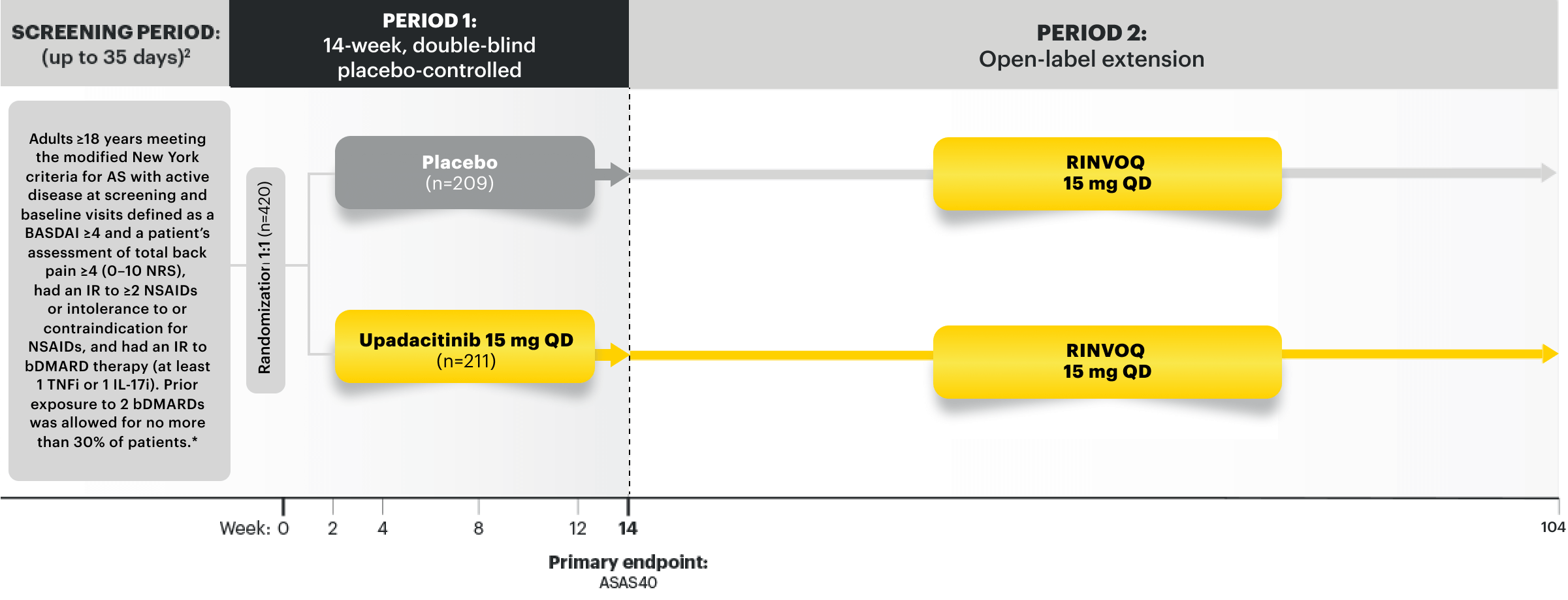
NRI-MI Data From SELECT-AXIS 2 Study 1: AS (bDMARD-IR)1,2
RINVOQ 15 mg (n=211), placebo (n=209)
ASAS40 | PRIMARY ENDPOINT
45%* RINVOQ vs 18% placebo at Week 14
*P<0.0001.2
SELECT-AXIS 2 Study 1: AS Design Intro1,2: 14-week, double-blind, parallel-group, placebo-controlled, Phase 3 study of 420 patients with active AS who had an intolerance or inadequate response to at least 2 NSAIDs and bDMARDs. Patients were randomized to receive RINVOQ 15 mg once daily or placebo. Patients could continue background NSAIDs. The primary endpoint was the proportion of patients achieving ASAS40 response at Week 14 vs placebo.
Please see Important Safety Information, including BOXED WARNING on Serious Infections, Mortality, Malignancies, Major Adverse Cardiovascular Events, and Thrombosis, below.
MMRM Data From SELECT-AXIS 2 Study 1: AS (bDMARD-IR)4
Mean Change
in hsCRP* (mg/mL)
-10.9† RINVOQ (n=202) vs
0.4 placebo (n=198) at Week 14
Mean Change in Peripheral
Joint Pain and Swelling* (NRS 0–10)
-2.32† RINVOQ (n=211) vs
-1.17 placebo (n=209) at Week 14
Mean Change
in PtGA Disease Activity* (NRS 0–10)
-2.97† RINVOQ (n=211) vs
-1.38 placebo (n=209) at Week 14
Mean Change in
Duration of Morning Stiffness* (NRS 0–10)
-2.80† RINVOQ (n=211) vs
-1.54 placebo (n=209) at Week 14
Mean Change
in Total Back Pain‡ (NRS 0–10)
-3.00§ RINVOQ (n=211) vs
-1.47 placebo (n=209) at Week 14
Mean Change
in MASES‡||
-2.6§ RINVOQ (n=148) vs
-1.1 placebo (n=162) at Week 14
*DATA LIMITATIONS: Data labeled as a primary or ranked secondary endpoint were multiplicity-controlled for comparisons. All other comparisons were not adjusted for multiplicity; statistical significance has not been established.
*Select prespecified nonranked endpoint.
†P<0.0001; analyses were not controlled for multiplicity.
‡Ranked secondary endpoint.
§P<0.0001.
||For patients with baseline presence of enthesitis (MASES>0).
SELECT-AXIS 2 Study 1: bDMARD-IR patients5,6
ALL DATA ARE AS OBSERVED

76%#
improvement in high
sensitivity C-reactive protein
at 2 years (OLE) (n=170)

PERIPHERAL JOINTS¶
64%
improvement in peripheral
joint pain and swelling at
2 years (OLE) (n=166)

PtGA DISEASE ACTIVITY¶
69%
improvement in patient global
assessment of disease activity
at 2 years (OLE) (n=168)


STIFFNESS¶
63%
improvement in duration
of morning stiffness at
2 years (OLE) (n=168)

PAIN¶
67%
improvement in total
back pain at 2 years (OLE)
(n=168)

ENTHESITIS¶
64%
of AS patients achieved
complete resolution of
enthesitis (MASES=0||) at
2 years (OLE) (n=125)

INFLAMMATION¶
76%#
improvement in high
sensitivity C-reactive protein
at 2 years (OLE) (n=170)
PERIPHERAL JOINTS¶
64%
improvement in peripheral
joint pain and swelling at
2 years (OLE) (n=166)
PtGA DISEASE ACTIVITY¶
69%
improvement in patient global
assessment of disease activity
at 2 years (OLE) (n=168)

STIFFNESS¶
63%
improvement in duration
of morning stiffness at
2 years (OLE) (n=168)
PAIN¶
67%
improvement in total
back pain at 2 years (OLE)
(n=168)
ENTHESITIS¶
64%
of AS patients achieved
complete resolution of
enthesitis (MASES=0||) at
2 years (OLE) (n=125)
¶DATA LIMITATIONS: Outcome measures were prespecified to be collected as continuous outcomes. Percentage improvement in continuous outcomes were not prespecified. No statistical or clinical conclusions can be made.
#Value represents the median improvement from baseline.
As Observed (AO) analysis: Patients with missing data at a specific time are not included, which may enrich the population and increase the response rates.
OLE LIMITATIONS: There is potential for enrichment of OLE data; unblinding patients may cause bias related to the overall treatment effect.
NRI-MI Data From SELECT-AXIS 2 Study 1: AS2,3
RINVOQ 15 mg (n=211), placebo (n=209)
ASDAS-CRP LDA | RANKED SECONDARY ENDPOINT
44%* RINVOQ vs 10% placebo at Week 14
INACTIVE DISEASE ACTIVITY | RANKED SECONDARY ENDPOINT
13%* RINVOQ vs 2% placebo at Week 14
*P<0.0001.
Almost 3 Out of 4 (71%) RINVOQ Patients Achieved ASDAS-CRP LDA at 2 Years7
SELECT-AXIS 2 Study 1:
bDMARD-IR patients
ALL DATA ARE AS OBSERVED

BASED ON A POST HOC ANALYSIS:
In patients who achieved ASDAS-CRP LDA at Weeks 14 and 52 (n=66/211),

MAINTAINED LDA RESPONSE
AT 2 YEARS (OLE) (AO)8*
*DATA LIMITATIONS: Post hoc analysis was not included as a prespecified ranked endpoint or controlled for multiplicity. Therefore, no clinical or statistical conclusions can be drawn.
As Observed (AO) analysis: Patients with missing data at a specific time are not included, which may enrich the population and increase the response rates.
OLE LIMITATIONS: There is potential for enrichment of OLE data; unblinding patients may cause bias related to the overall treatment effect.
SAFETY CONSIDERATIONS
Serious Infections: RINVOQ-treated patients are at increased risk of serious bacterial (including tuberculosis [TB]), fungal, viral, and opportunistic infections leading to hospitalization or death. Most patients who developed these infections were taking concomitant immunosuppressants, such as methotrexate or corticosteroids.
Mortality: A higher rate of all-cause mortality, including sudden cardiovascular (CV) death, was observed with a Janus kinase inhibitor (JAKi) in a study comparing another JAKi with tumor necrosis factor (TNF) blockers in rheumatoid arthritis (RA) patients ≥50 years with ≥1 CV risk factor.
Malignancies: Malignancies have occurred in RINVOQ-treated patients. A higher rate of lymphomas and lung cancer (in current or past smokers) was observed with another JAKi when compared with TNF blockers in RA patients.
Major Adverse Cardiovascular Events: A higher rate of CV death, myocardial infarction, and stroke was observed with a JAKi in a study comparing another JAKi with TNF blockers in RA patients ≥50 years with ≥1 CV risk factor. History of smoking increases risk.
Thromboses: Deep venous thrombosis, pulmonary embolism, and arterial thrombosis have occurred in patients treated for inflammatory conditions with JAK inhibitors, including RINVOQ. A higher rate of thrombosis was observed with another JAKi when compared with TNF blockers in RA patients.
Hypersensitivity: RINVOQ is contraindicated in patients with hypersensitivity to RINVOQ or its excipients.
Other Serious Adverse Reactions: Hypersensitivity Reactions, Gastrointestinal Perforations, Laboratory Abnormalities, and Embryo-Fetal Toxicity.
Achieving ASDAS-Low Disease Activity May Indicate Disease Control, Making It a Measure to Strive for in Patients With AS
Distribution of RINVOQ Patients by ASDAS-CRP State at Baseline and Week 104
SELECT-AXIS 2 Study 1: bDMARD-IR patients3,9
ALL DATA ARE AS OBSERVED |
Open-Label Extension (OLE)

Improvement in ASDAS-CRP Score Seen as Early as Week 14
MMRM Data From SELECT-AXIS 2 Study 1: AS2
RINVOQ 15 mg (n=211), placebo (n=209)
Mean Change in ASDAS-CRP Score |
RANKED SECONDARY ENDPOINT
-1.52* RINVOQ vs -0.49 placebo at Week 14
*P<0.0001.
The ASDAS-CRP score is a composite measure that assesses common patient symptoms and objective signs of inflammation. The score is calculated by assessing the following components on a numerical rating scale (from 0 to 10)10 or blood serum level (mg/L).
ASDAS-CRP Disease Activity Score Components at 2 Years
SELECT-AXIS 2 Study 1:
bDMARD-IR patients11,12
ALL DATA ARE AS OBSERVED |
Open-Label Extension (OLE)

DATA LIMITATIONS: ASDAS component analyses were prespecified nonranked endpoints not controlled for multiplicity. Therefore, no clinical or statistical conclusions can be drawn.
As Observed (AO) analysis: Patients with missing data at a specific time are not included, which may enrich the population and increase the response rates.
OLE LIMITATIONS: There is potential for enrichment of OLE data; unblinding patients may cause bias related to the overall treatment effect.
Improvement in Total Back Pain and Nocturnal Back Pain
Ranked Secondary Endpoint at Week 14 with Response Rates Out to 2 Years
SELECT-AXIS 2 STUDY 1: bDMARD-IR patients2,3
ΔTotal Back Pain from baseline, MMRM*

*Total back pain defined on a numeric rating scale (0–10) based on the following question, "What is the amount of back pain that you experienced at any time during the last week?"2
†P<0.0001.2

improvement (n=206) vs 19% (n=203) with placebo at Week 14 as observed13 and

improvement (n=168) at 2 years as observed3
DATA LIMITATIONS: Data labeled as a primary or ranked secondary endpoint at Week 14 were multiplicity-controlled. All other comparisons were not adjusted for multiplicity; statistical significance has not been established.
As Observed (AO) analysis: Patients with missing data at a specific time are not included, which may enrich the population and increase the response rates.
OLE LIMITATIONS: There is potential for enrichment of OLE data; unblinding patients may cause bias related to overall treatment effect.
Ranked Secondary Endpoint at Week 14 with Response Rates Out to 2 Years
SELECT-AXIS 2 STUDY 1: bDMARD-IR patients2,3
ΔNocturnal Pain from baseline, MMRM*

*Nocturnal back pain defined on a numeric rating scale (0–10) based on the following question, “What is the amount of back pain at night that you experienced during the last week?”2
†P<0.001.4

improvement (n=205) vs 21% (n=202) with placebo at Week 14 as observed13 and

improvement (n=211) at 2 years as observed14
DATA LIMITATIONS: Data labeled as a primary or ranked secondary endpoint at Week 14 were multiplicity-controlled. All other comparisons were not adjusted for multiplicity; statistical significance has not been established.
As Observed (AO) analysis: Patients with missing data at a specific time are not included, which may enrich the population and increase the response rates.
OLE LIMITATIONS: There is potential for enrichment of OLE data; unblinding patients may cause bias related to overall treatment effect.
Please see Important Safety Information, including BOXED WARNING on Serious Infections, Mortality, Malignancies, Major Adverse Cardiovascular Events, and Thrombosis, below.
Durable Rates of ASAS40 Response Up to 2 Years1-3
82% of RINVOQ AS Patients Achieved AS at 2 Years3
SELECT-AXIS 2 Study 1:
bDMARD-IR patients
ALL DATA ARE AS OBSERVED

*Total back pain defined on an NRS (0–10) based on the following question: "What is the amount of back pain that you experienced at any time during the last week?"2
†Defined as mean of BASDAI questions 5 and 6.
ASAS40 reflects a ≥40% improvement in 3 of the 4 following domains with no worsening in the fourth domain2:
ASAS Domains
- Total back pain*
- Inflammation (morning stiffness)†
- Physical function (BASFI)
- Patient global assessment of disease activity
As Observed (AO) analysis: Patients with missing data at a specific time are not included, which may enrich the population and increase the response rates.
OLE LIMITATIONS: There is potential for enrichment of OLE data; unblinding patients may cause bias related to the overall treatment effect.
Interested in the safety data for RINVOQ?
See RINVOQ’s safety data across clinical trials
Explore RINVOQ data in gastroenterology
For moderate to severe Crohn's disease (CD) or moderate to severe ulcerative colitis (UC) in adult TNFi-IR patients1